

The assembly algorithms that have been developed so far intend to provide better assemblies evaluated under different criteria. Hence, depending on the specific scenario the assembly process might produce better results if we use the most appropriate assembler. Even though contiguous genomes may not be produced, segments from the reference genomes can be obtained using existing assembly methods. Therefore, the need to evaluate the quality of assemblies exists. These evaluations help researchers to pick different assemblers for different scenarios.
How can we know whether the assemblies we obtain from reads using currently available assemblers are correct or not? In this article, we will see how to determine the quality of assemblies using QUAST, which is one of the most famous assessment tools available for genome assemblies. Let’s get started.
What is QUAST?
QUAST stands for QUality Assessment Tool. QUAST can evaluate assemblies using reference genomes, as well as without reference genomes. QUAST produces detailed reports, tables and plots which show the different aspects of assemblies.
Download QUAST
You can go to the official website of QUAST and click on the DOWNLOAD button.
You will be directed to a SOURCEFORGE download page from where you can download the latest version (quast-5.0.2 when I was writing this article) of QUAST. The pre-compiled binaries will be downloaded and you can run it straight away after extracting.
tar -xf quast-5.0.2.tar.gz
cd quast-5.0.2
quast.pyYou can see the following after executing quast.py or python quast.py.
QUAST: Quality Assessment Tool for Genome Assemblies
Version: 5.0.2Usage: python quast.py [options] <files_with_contigs>Options:
-o --output-dir <dirname> Directory to store all result files [default: quast_results/results_<datetime>]
-r <filename> Reference genome file
-g --features [type:]<filename> File with genomic feature coordinates in the reference (GFF, BED, NCBI or TXT)
Optional 'type' can be specified for extracting only a specific feature type from GFF
-m --min-contig <int> Lower threshold for contig length [default: 500]
-t --threads <int> Maximum number of threads [default: 25% of CPUs]These are basic options. To see the full list, use --helpOnline QUAST manual is available at http://quast.sf.net/manual
Once you have ensured that QUAST is running correctly, we can start to assess some assemblies.
Obtaining an Example Assembly
We will be using the example dataset used in the Flye assembler. The example dataset consists of reads of an E. coli genome (Escherichia coli str. K-12 substr. MG1655 with NCBI accession number CP009685). The reads consist of PacBio reads.
You can download the dataset with reads using the following command.
wget https://zenodo.org/record/1172816/files/E.coli_PacBio_40x.fastaLet’s assemble this dataset using the Flye assembler.
flye --pacbio-raw E.coli_PacBio_40x.fasta --out-dir my_assembly --threads 8Now we have an example assembly. The contigs of the final assembly can be found in the file assembly.fasta. Let’s see how good the quality of the assembly is.
Using QUAST
You can run QUAST by providing the contigs file containing the final assembly and the reference genome.
quast.py my_assembly/assembly.fasta -r ref.fasta -o quastResultNow you can view the final report from the report.html file in the output folder.
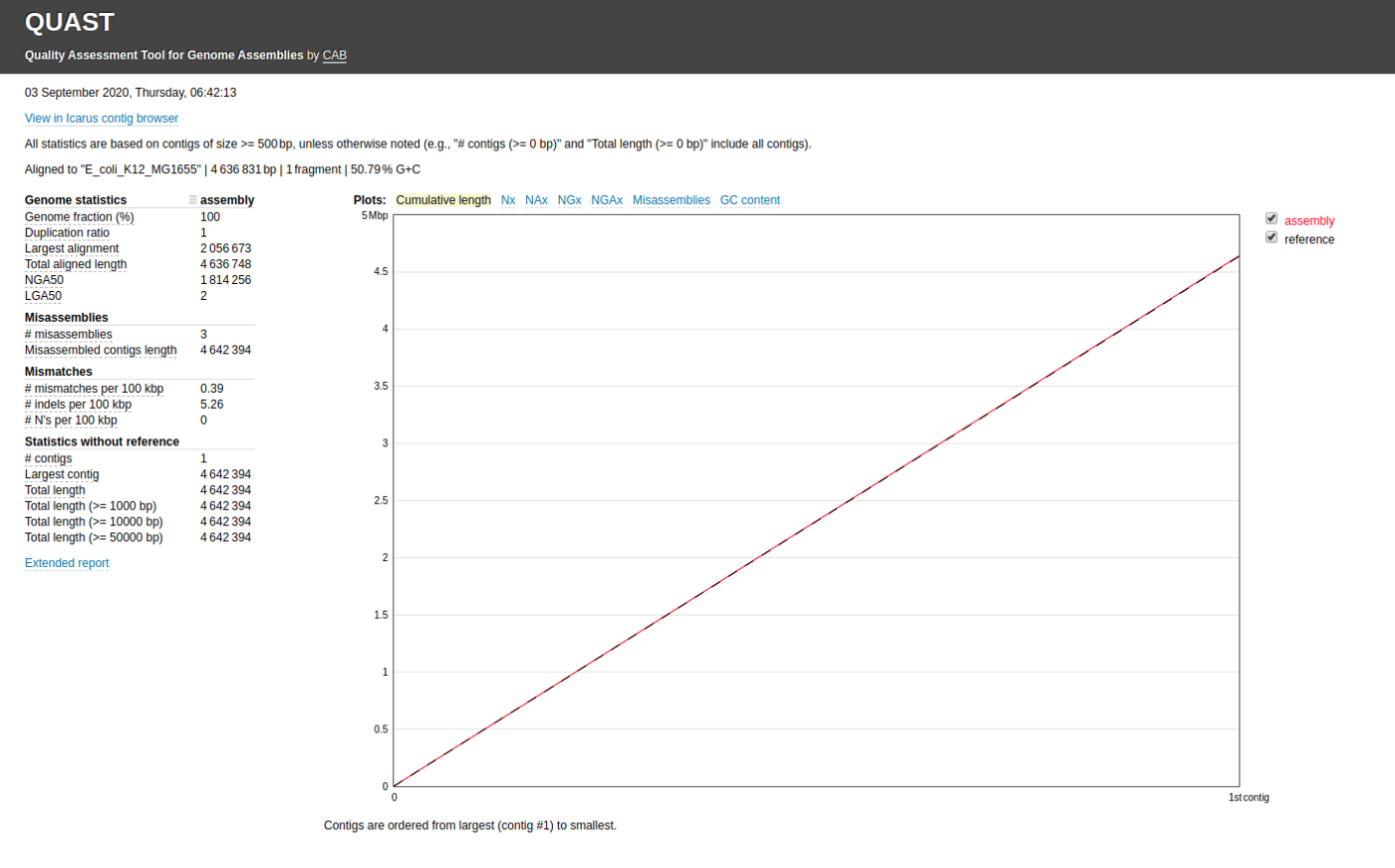
You can also compare multiple assemblies (assemly1.fasta and assembly2.fasta) as shown. You can specify labels for each assembly as well.
quast.py assemly1.fasta assembly2.fasta -l label1,label2 -r ref.fasta -o quastResult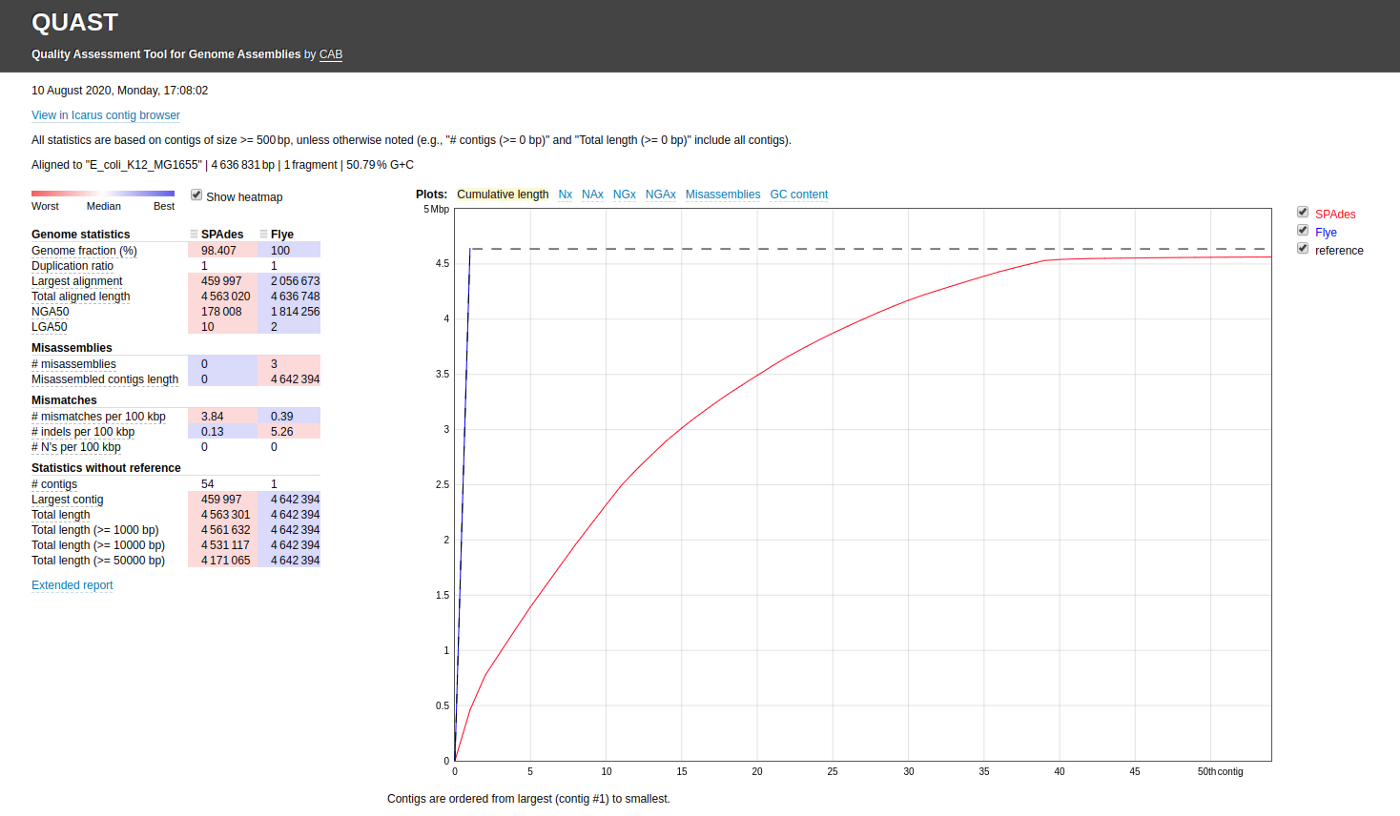
You can note the following common evaluation measures that are used to assess the quality of genomes.
- Genome fraction
- Largest alignment
- NGA50
- LGA50
- Number of misassemblies
- Number of contigs
QUAST provides sample explanations for each of these measures. You can hover over each measure and a popup message will be shown with the explanation.
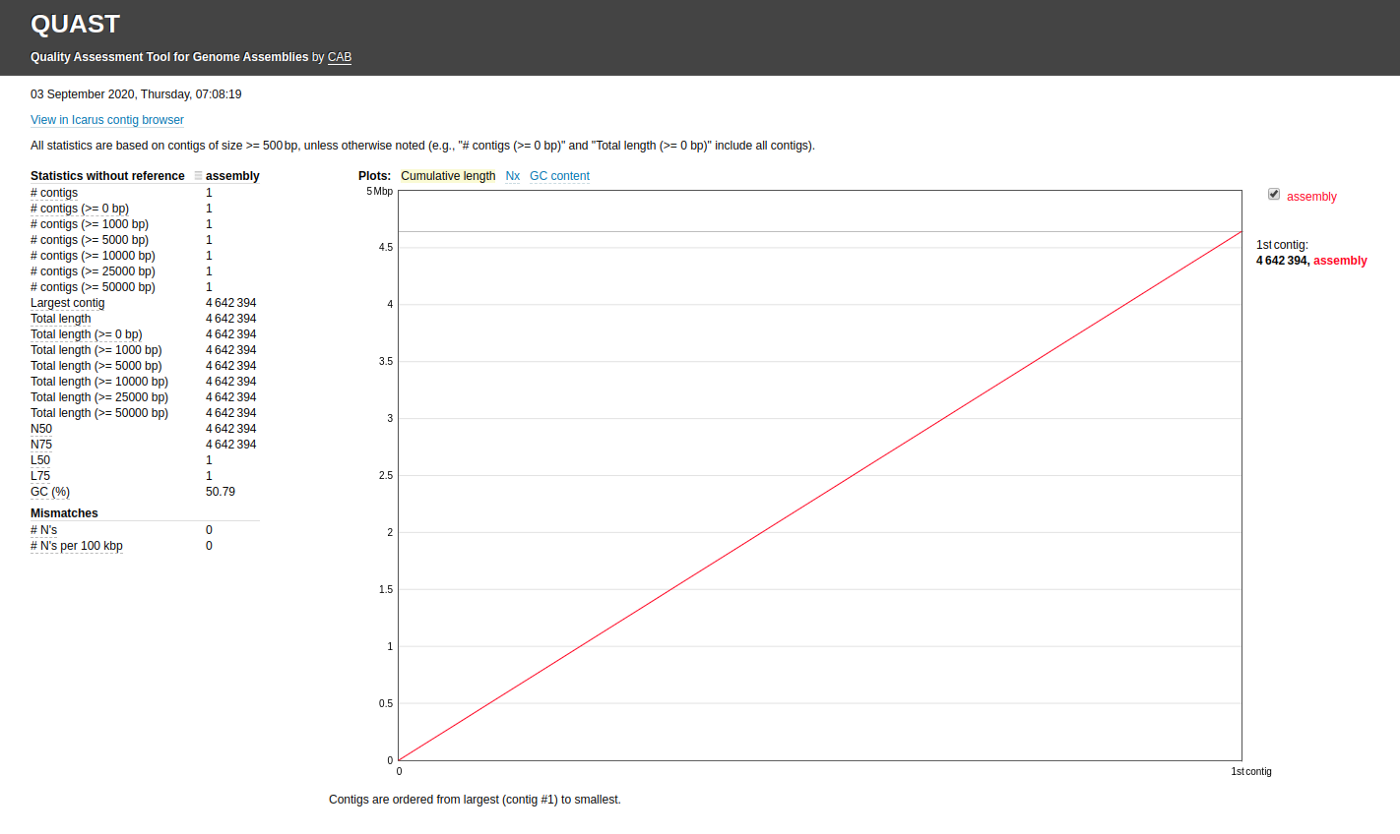
You can also assess your assembly without providing any reference genomes.
quast.py my_assembly/assembly.fasta -o quastResultYour result will contain details of the statistics without any references such as,
- Number of contigs
- Largest contig
- Total length
- N50
- L50
Icarus Contig Browser
Icarus is a tool available within QUAST which can visualise assemblies for analytical purposes.

You can view how well your assembly aligns with the reference genome.
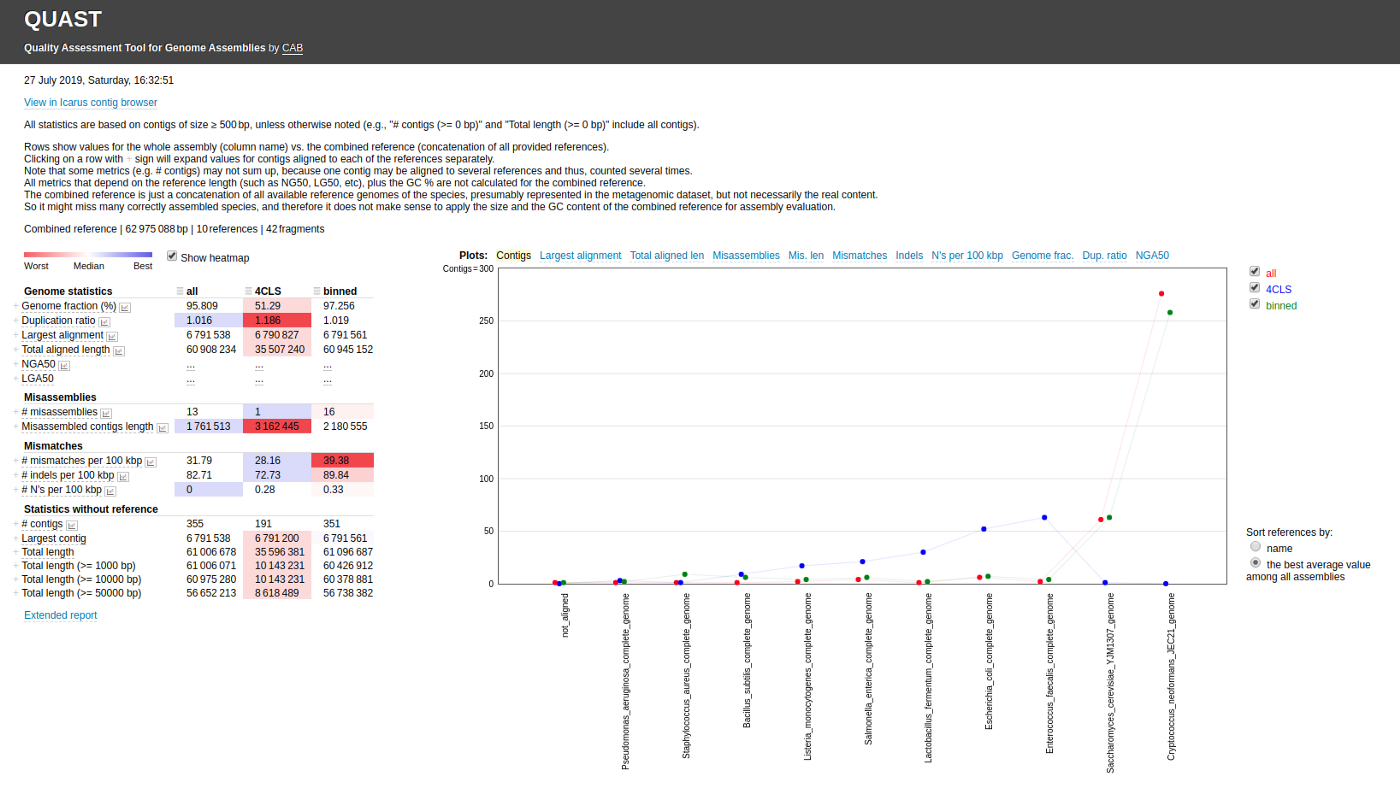
MetaQUAST: QUAST for Metagenomics Assemblies
QUAST provides a version named MetaQUAST, that allows us to assess metagenomics assemblies. You can provide multiple assemblies and compare them at once. Moreover, you can provide multiple reference genomes as well.
You can run MetaQUAST as follows.
metaquast.py meta.contigs1.fasta meta.contigs2.fasta -l label1,label2 -R References/ -t 8 -o metaquastResultSimilar to QUAST, you can provide labels for each assembly so that they will be displayed in the final report. Moreover, you can provide a single folder containing all the reference genomes for the assessment.
Final Thoughts
Hope you found this article useful and informative as a starting point for using quality assessment tools for genome assemblies. Feel free to use these tools for your projects and research work as they are freely available.
Cheers, and stay safe!
This article was originally published in The Computational Biology Magazine on Medium.
You can find the original article at https://medium.com/computational-biology/assessing-the-quality-of-genome-assemblies-using-quast-94fec3f8cb70
