

Have you ever wondered how life formed from the primordial soup and evolved to the different life forms which can be seen at present? How did different species evolved from their ancestors and what relationships do they have with each other? The answers to these questions can be answered through the study of phylogenetics.
This article will walk you through the following topics.
- What is Phylogenetics?
- Phylogenetic Trees
- Algorithms used for Phylogenetic Inference
- Sample Practice Task with Code
What is Phylogenetics?
According to Wikipedia,
Phylogenetics is the study of the evolutionary history and relationships among individuals or groups of organisms.
The relationships among organisms are discovered through phylogenetic inference methods where heritable traits, such as DNA sequences or morphologies can be observed under a certain model of evolution. The result of these analyses is a phylogeny (also known as a phylogenetic tree). It is a diagram depicting a hypothesis about the history of the evolutionary relationships of a group of organisms or a family of genes.
Molecular phylogenetics is a branch of phylogenetics that analyses how certain molecules, mainly DNA sequences and proteins have changed over time, to determine evolutionary relationships of a group of organisms or a family of genes.
Phylogenetic Trees
Phylogenetic trees represent evolutionary relationships between organisms or genes. The pattern of branching in a phylogenetic tree reflects how species or other groups have evolved from a series of common ancestors. An example of a phylogenetic tree is the Tree of Life which denotes how various species of organisms have evolved since the birth of Earth.
In a phylogenetic tree, the species or groups of interest are found at the tips of lines known as branches. The points where branches are divided are called branch points.
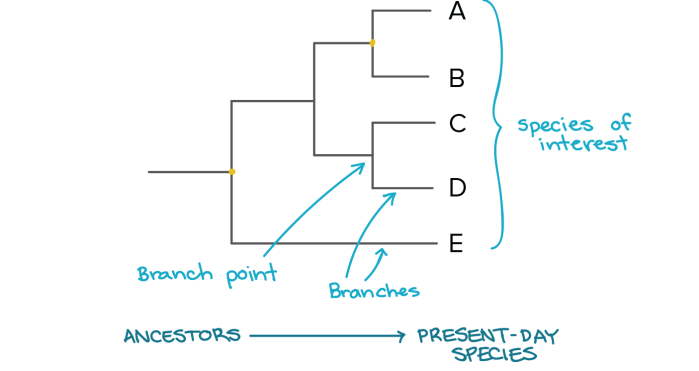 Image Source: Khan Academy
Image Source: Khan Academy
Two species are more related if they have a more recent common ancestor and less related if they have a less recent common ancestor.
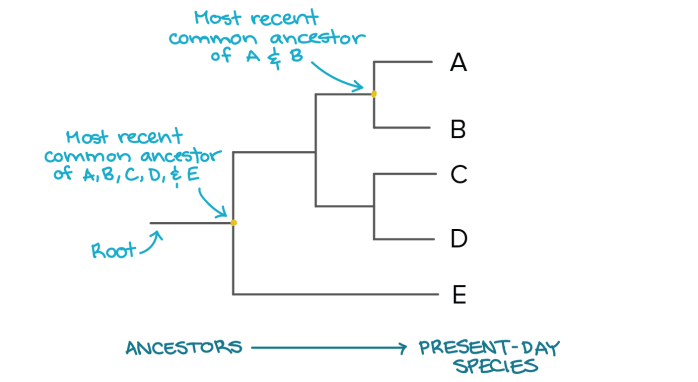 Image Source: Khan Academy
Image Source: Khan Academy
Given below is a phylogenetic tree for primates based on their genetic data. Gorillas and Orangutans have diverged earlier than other primate groups. The Homo lineage (humans) has moved along one path where as the Pan lineage has moved along another path. Later on, the Pan lineage has divided, yielding Chimpanzees and Bonobos.
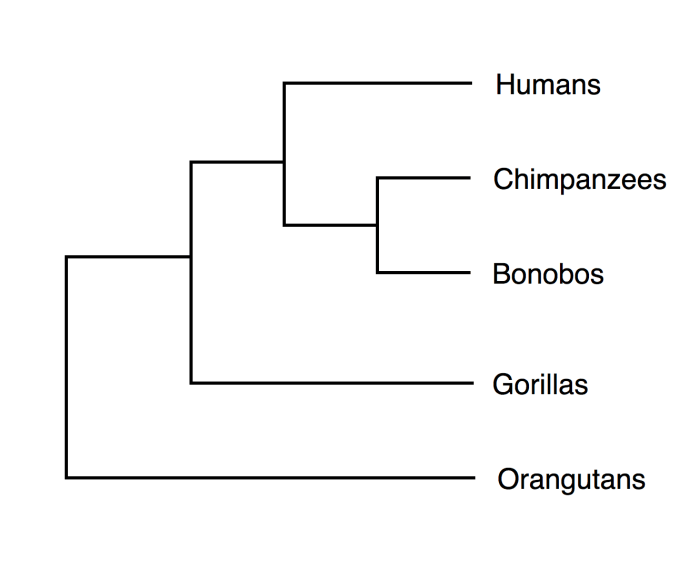 Phylogenetic tree for primates based on their genetic data
Phylogenetic tree for primates based on their genetic data
Algorithms used for Phylogenetic Inference
There are three main categories of algorithms that are used for phylogenetic inference from any type of biological data. They are,
- Distance-based methods
- Maximum Parsimony (MP) methods
- Probabilistic methods
1. Distance-based Methods
Distance-based methods compute an evolutionary distance, which is the number of changes that have occurred for two species considered to diverge from a common ancestor. However, these methods face problems with accuracy when it comes to dealing with large volumes of data which have very distant relationships.
2. Maximum Parsimony (MP) methods
MP methods infer a tree that minimizes the total number of changes, known as mutations, required to explain the data. Under the maximum parsimony criterion, the shortest possible tree that explains the data is considered as the best tree. This best tree is known as the most-parsimonious tree. Heuristic search is performed to quickly generate the most-parsimonious tree. Since this methods considers the shortest possible tree as the best tree, actual evolutionary changes that have occurred may be underestimated.
3. Probabilistic methods
Probabilistic methods, such as Maximum Likelihood (ML) and Bayesian inference, attempt to find a tree that maximizes the conditional or posterior probability of observing the data. Phylogenetic studies at present, widely utilize Bayesian frameworks due to the possibility of account for the phylogenetic uncertainty, availability of efficient algorithms and their implementation as various computer programs.
Bio.Phylo — Time to Practice
Since we have a basic idea about phylogenetic trees, it is time to try out some coding. I have introduced a set of Python tools named Biopython in one of my previous articles, which can be used to analyze biological data. If you haven’t gone through it make sure to check it out as well.
I will be using the Bio.Phylo module which provides classes, functions and I/O support for working with phylogenetic trees. You can go through the official documentation to get more details about this module.
Task — Construct the phylogenetic tree for the given DNA sequences
Consider you are provided five DNA sequences with their labels in the beginning of each line. You can find these sequences in a file named as msa.phy in the official biopython test material for tree construction. The sequences considered are given below.
- Alpha AACGTGGCCACAT
- Beta AAGGTCGCCACAC
- Gamma CAGTTCGCCACAA
- Delta GAGATTTCCGCCT
- Epsilon GAGATCTCCGCCC
We are given the task of constructing the phylogenetic tree to represent these sequences based on distance-based phylogenetic inference methods.
Currently, Bio.Phylo module has two types of tree constructors: DistanceTreeConstructor and ParsimonyTreeConstructor. We will be using DistanceTreeConstructor for this task.
Furthermore, the DistanceTreeConstructor supports two heuristic algorithms: UPGMA (Unweighted Pair Group Method with Arithmetic Mean) and NJ (Neighbor Joining). We will be using the UPGMA algorithm. You can read more about the UPGMA algorithm from this link.
Solution
Firstly, make sure you have downloaded the msa.phy file which contains the input sequences and include it in your current working directory.
Given below is the python code to create the phylogenetic tree for the given DNA sequences. Note how we have used Bio.Phylo module and its functionality.
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| # Import modules | |
| from Bio import Phylo | |
| from Bio.Phylo.TreeConstruction import DistanceCalculator | |
| from Bio.Phylo.TreeConstruction import DistanceTreeConstructor | |
| from Bio import AlignIO | |
| # Read the sequences and align | |
| aln = AlignIO.read('msa.phy', 'phylip') | |
| # Print the alignment | |
| print aln | |
| # Calculate the distance matrix | |
| calculator = DistanceCalculator('identity') | |
| dm = calculator.get_distance(aln) | |
| # Print the distance Matrix | |
| print('\nDistance Matrix\n===================') | |
| print(dm) | |
| # Construct the phylogenetic tree using UPGMA algorithm | |
| constructor = DistanceTreeConstructor() | |
| tree = constructor.upgma(dm) | |
| # Draw the phylogenetic tree | |
| Phylo.draw(tree) | |
| # Print the phylogenetic tree in the terminal | |
| print('\nPhylogenetic Tree\n===================') | |
| Phylo.draw_ascii(tree) | |
By running the code, we can get the phylogenetic tree as a graphical visualization as well as get it printed in the terminal as shown below.
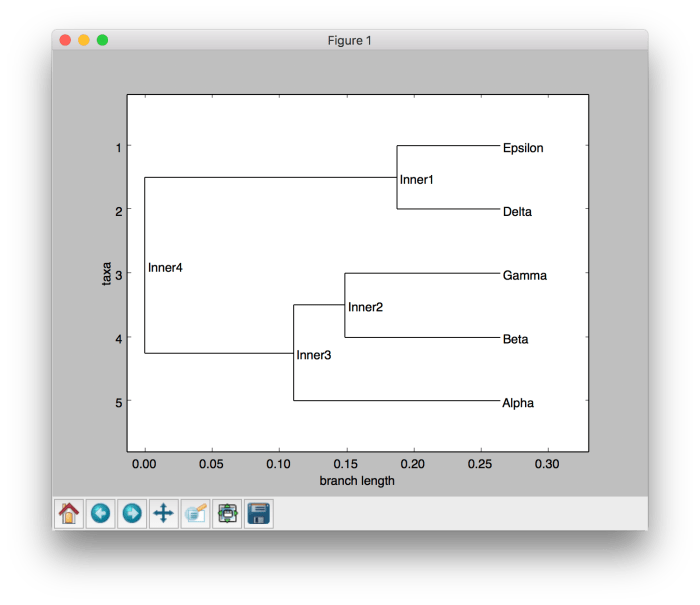 Graphical visualization of the phylogenetic tree using UPGMA
Graphical visualization of the phylogenetic tree using UPGMA
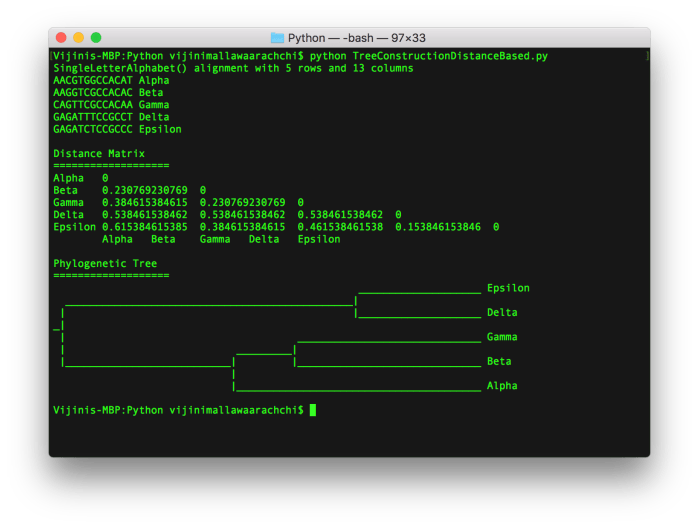 The phylogenetic tree using UPGMA printed in the terminal at the end
The phylogenetic tree using UPGMA printed in the terminal at the end
If you use NJ algorithm instead of UPGMA algorithm, the resulting tree will be changed as shown below.
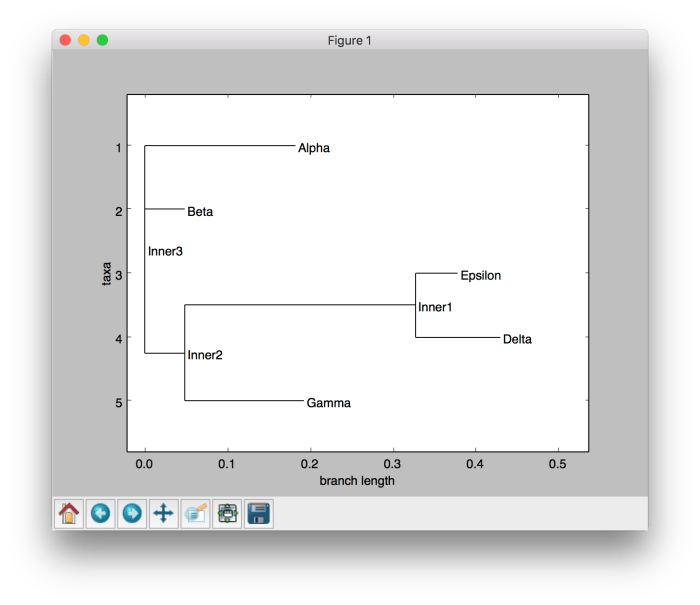 Graphical visualization of the phylogenetic tree using NJ
Graphical visualization of the phylogenetic tree using NJ
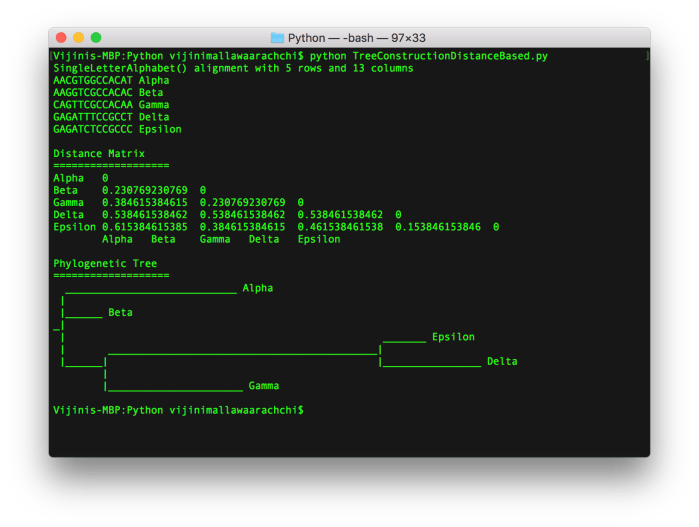 The phylogenetic tree using NJ printed in the terminal at the end
The phylogenetic tree using NJ printed in the terminal at the end
Hope you enjoyed reading this article and learned useful and interesting things about molecular genetics and how to use Biopython to construct phylogenetic trees from a given set of sequences. I would love to hear your thoughts and ideas.
Thanks for reading… 😊
Originally posted in Medium.com at Towards Data Science

Loved it! In the future I will have to learn and understand more deeply how these programs function.
Best wishes from Brazil
LikeLike
Thank you very much! 😀
LikeLike